



Маленький Фаддей из Архангельской обрасти — младший в семье, у него есть еще старшая сестра и совсем взрослый брат. Оба они здоровы, а вот Фаддей всю свою пока еще короткую жизнь борется с генетическим заболеванием, которое даже выговорить сложно: дефицит декарбоксилазы ароматических аминокислот. Проще говоря, из-за ряда мутаций, нарушается выработка фермента, который в свою очередь вызывает дефицит дофамина и серотонина. Как говорят в «Круге добра» — государственном фонде, созданном для поддержки детей с тяжелыми, в том числе редкими (орфанными) заболеваниями, в мировой литературе описано порядка 180 случаев такой болезни.
При этом болезнь долго «маскируется» под ДЦП, поэтому поставить правильный диагноз непросто. Вот и Фаддею в 1,5 года после долгого мыкания по больницам диагностировали детский церебральный паралич. И только в начале 2022 года, после множества исследований, ряда госпитализаций и безуспешных попыток реабилитаций, по результатам нового генетического анализа наконец-то добрались до истинной причины его недуга.
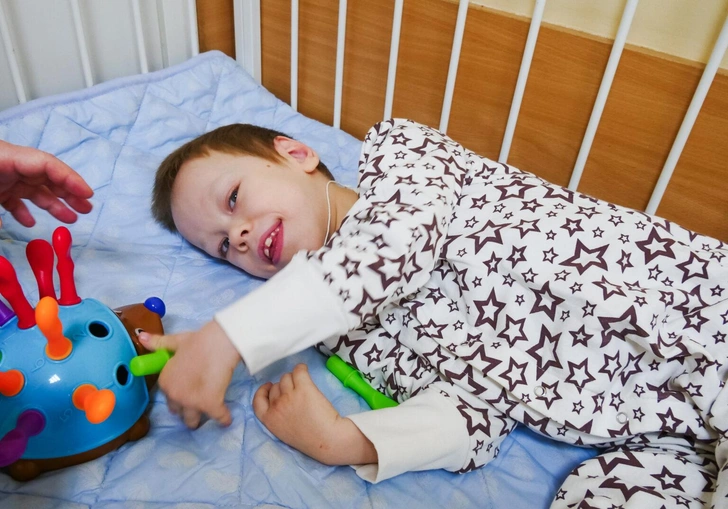
«Сын рос гораздо медленнее сверстников, была на лицо серьезная задержка психомоторного развития, — говорит мама мальчика Надежда Александрова. — Взгляд у него был осознанный, нас — родителей, брата и сестру — вроде бы узнавал, однако физически имел много ограничений. Фаддей только лежал, не ползал, не держал вертикально голову, не мог переворачиваться, не пытался сам взять ложку, хотя с аппетитом ел густую протертую кашицу».
- Источник:
Фото: РДКБ/Круг Добра
После того, как лечение скорректировали, в развитии Фаддея наметился явный прогресс: он начал переворачиваться, улучшалась моторика — мальчик.без труда встает на колени, тянет руку, чтобы поздороваться, появился некий социальный контакт. Но и родители, и врачи, понимали, что болезнь опережает их.
«Уже в раннем возрасте болезнь проявляется тяжелыми симптомами: мышечная гипотония, задержка двигательного и психоречевого развития, расстройство вегетативной системы, — объясняет профессор, доктор медицинских наук., заведующая отделением медицинской генетики Российской детской клинической больницы Светлана Михайлова. —. Впервые в России диагноз был поставлен двум детям в отделении медицинской генетики Российской детской клинической больницы в 2012 году, и до недавнего времени лечения не существовало».
Сейчас препарат есть — новейшая разработка иностранных ученых Эладокаген экзупарвовек («Апстаза») позволяет восстановить выработку фермента за счет внесения в организм здоровой копии гена. Сложно говорить о том, что случится чудо, и ребенок полностью восстановится, но улучшения после введения лекарства наступают очень быстро: есть шанс, что ребенок научится сам сидеть, держать голову.
Но цена… Апстаза — один из самых дорогостоящих препаратов в мире, его стоимость около 3 миллионов евро. Собрать такую сумму ни одной семье практически не по силам. Но Фаддей — первым в России — получил жизненно важный укол: лекарство закупил Фонд «Круг добра», а уникальную операцию по введению препарата прямо в головной мозг ребенка провели нейрохирурги Российской детской клинической больницы Минздрава России. Россия стала шестой страной в мире, где начали применять этот препарат.
Кстати, сама операция тоже уникальна: лекарство надо «доставить» в строго определенные участки мозга микродозами. При этом время ограничено: препарат транспортируют при температуре — 70 градусов, размораживают за полчаса до операции и хранится он после этого не более 6 часов. Поэтому хирургам пришлось провести и масштабную подготовку: с помощью 3D-моделирования построить 4 траектории для введения препарата.
«Сразу после успешного введения в нужные зоны мозга начинается выработка нейромедиаторов, что позволяет остановить патологический процесс утраты двигательных функций и нарушений развития у пациента, — говорит Светлана Михайлова. — Ожидаем возвращения к ребенку контроля над движениями, возможности садиться, самостоятельно передвигаться».
А тем временем нейрохирурги РДКБ готовятся к еще одной такой операции. Как рассказали в фонде, в ближайшие дни уникальную терапию должна получить 7-летняя Варвара из Краснодарского края.
